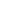更新时间:2016-05-12 11:04 浏览: 次 作者:admin
疾病研究的实质性进展取决于能够准确检测基因组畸变,并可以让人们对这些信息做出快速可靠解释的分析工具。新一代测序技术相对传统的体外诊断方法,有卓越的稳定性和可靠性。未来新一代测序技术的研究将采用更大的样本群,提供统计学上可靠的关联,并逐步构建完整的突变数据库,使得医生能够为患者提供更适合的治疗方法,促进个性化医疗时代的到来。
基因组学研究——全基因组重测序分析
在已有人类全基因组参考序列及其基因功能注释的基础上,对人类基因组DNA 样本进行重测序,寻找不同个体基因组相对于人类参考基因组序列的差异,揭示基因组中的遗传变异对于表型多样性和疾病易感性差异的影响,推断群体迁徙及进化的历史过程。
1.应用领域
通过高通量测序识别发现de novo 的somatic 和germ line 突变,结构变异SV,体细胞突变SNV,拷贝数变异CNV 以及SNP 的座位等;在比较基因组学,群体遗传学和药物基因组学的综合层面上,深入探索疾病基因组和癌症基因组的致病机理,药理机制及易感基因靶点。
2.技术路线
3.样品要求
样品类型:无降解,无污染的DNA 样品
样品总量:≥2.0 μg
样品浓度:≥30 ng/μl
4.测序样本量
建议癌组织和癌旁组织(或者血液)。
5.测序深度
个体人全基因重测序建议平均测序深度为30×;群体人全基因组重测序建议平均测序深度为4×-12×。
科普一下,测序深度(Sequencing Depth)是指测序得到的碱基总量(bp)与基因组大小(Genome)的比值,它是评价测序量的指标之一。测序深度与基因组覆盖度之间是一个正相关的关系,测序带来的错误率或假阳性结果会随着测序深度的提升而下降。重测序的个体,如果采用的是双末端或Mate-Pair方案,当测序深度在10~15X以上时,基因组覆盖度和测序错误率控制均得以保证。
6.案例:
双胞胎基因组中发现侵袭性癌症新线索
背景:由染色体重排为特征的急性白血病,通常需要结合其它分子水平的破坏才能发展为成熟的恶性肿瘤,但是他们之间的协作机制仍不了解。
结果:该研究通过全基因组测序分析了一对同卵双胞胎,一个患病,一个不患病,发现SETD2 基因存在双等位突变,并且在很多急性白血病患者中也发现存在能使该基因功能缺失的点突变,该突变也与多个主要的染色体异常有关。在
SETD2 基因突变的白血病幼稚细胞中,H3K36 三甲基化(H3K36me3)完全丧失。因此SETD2 可以作为肿瘤的抑制子,而SETD2-H3K36me3 通路的破坏是导致急性白血病的发展的一个明显表观遗传机制。
基因组学研究——外显子组测序分析
全外显子测序(Whole Genome Exome Sequencing, WES)指利用序列捕获技术将全基因组外显子区域DNA 捕捉,并富集后进行高通量测序的基因组分析方法,用于编码蛋白功能变异遗传突变的研究,外显子测序相对于基因组重测序成本较低,对研究已知基因的SNP、InDel 等具有较大的优势。
1.应用领域
应用于肿瘤,遗传疾病,复杂疾病等研究。深度测序能够发现常见和罕见突变,为治病机理和基因功能的研究提供重要技术支撑。
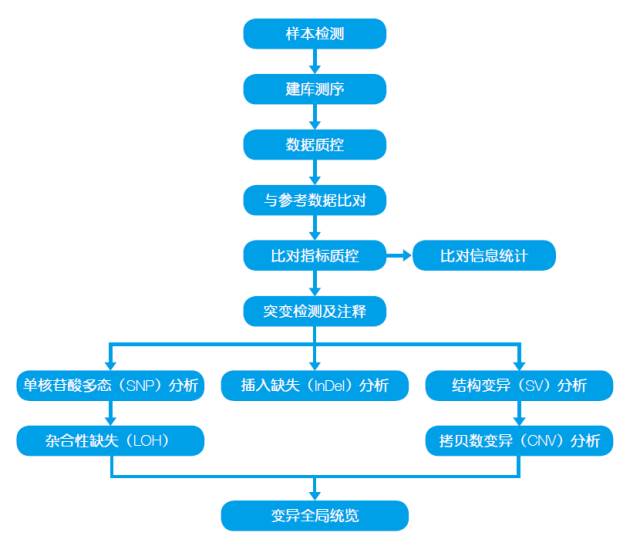
2.技术路线
3.样品要求
样品类型:无降解,无污染的DNA 样品
样品总量:≥2.0 μg
样品浓度:≥30 ng/μl
4.测序样本量
建议癌组织和癌旁组织(或者血液)至少10 对。
5.测序深度
肿瘤平均测序深度100× 以上;遗传,复杂疾病平均测序深度50× 左右。
6.案例:
目标区域测序与外显子组测序研究肿瘤异质性
背景:肿瘤异质性肿瘤的异质性是指肿瘤在经过多次分裂增殖后,其子细胞呈现出分子生物学或基因方面的改变,从而使肿瘤的侵袭能力、药物敏感性、预后等各方面产生差异。
结果:该研究以肺腺癌为研究对象,发现肿瘤异质性在不同癌症类型之间可能各不相同。研究人员对来自11 个手术切除局限性肺腺癌的48 个肿瘤区域进行了全外显子组测序,发现76% 的突变以及21 个已知癌症基因突变中的20 个存在于同一肿瘤的所有区域。平均21 个月后随访发现,11 位患者中有三人复发。这三人均具有更大比例的树枝突变(肿瘤某部分区域的异质性突变)占到了总突变的40%,而相比之下没有复发的患者中树枝突变只占17%。这意味着这些树枝突变有可能驱动了肿瘤的复发。
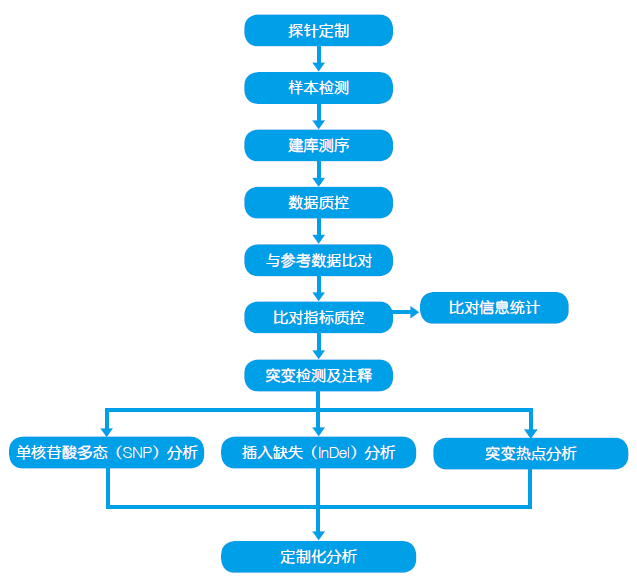
基因组学研究——目标区域测序分析
目标区域测序是指针对研究者感兴趣的基因组序列进行探针设计,然后进行目标区域捕获,利用新一代高通量测序平台进行分析的方法。该方法的特点在于,仅对基因组上的小部分区域进行测序,因此大大降低了测序成本。目标区域测序尤其适合于大样本量疾病样本分析,在已有研究基础的单基因病或复杂疾病致病基因研究中应用广泛。
1.应用领域
复杂疾病敏感位点分析,肿瘤易感基因检测,心脑血管疾病检测,用药指导基因检测,靶向药物筛选等,此外,目标区域测序还广泛应用于全基因组测序,GWAS 分析或连锁分析结果的验证。
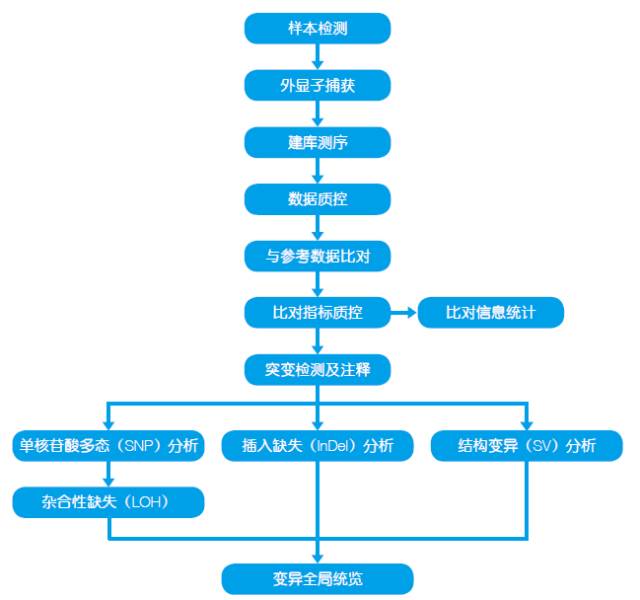
2.技术路线
3.样品建议
样品类型:无降解,无污染的DNA 样品
样品总量:≥2.0 μg
样品浓度:≥50 ng/μl
4.测序样本量
建议癌组织和癌旁组织(或者血液)至少10 对。
5.测序深度
遗传突变平均测序深度50×-100×;目标肿瘤平均测序深度100×-10000×;靶向药物筛选平均测序深度100×-10000×。
6.案例:
肿瘤目标区域测序分析循环肿瘤DNA
背景:循环肿瘤 DNA(circulating tumor DNA, ctDNA)是一类被认为具有广泛应用前景的肿瘤标志物,可用于肿瘤发展及预后状态的无创测定。最近发展出的癌症个体化深度测序分析方法(CAPP-Seq)利用定制化的 NimbleGen SeqCap EZ Choice Library 对样本进行靶向捕获后再进行深度测序,其对癌症的 ctDNA 检测结果灵敏度高,特异性强且经济可行。
结果:该研究针对非小细胞肺癌,设计覆盖了非小细胞肺癌95% 的突变位点的芯片,在 II-IV 期的肺癌患者的 ctDNA 中突变检出率为100%,在I 期患者中突变检出率为50%,并且可以检测出低至~0.02% 的特异性肿瘤突变。结合药物治疗后,通过影像学检测残余肿瘤的大小与 ctDNA 含量的关系,证明 ctDNA 的水平能够比影像学更早且更敏感的对肿瘤发展进行评估,因此CAPP-Seq 的方法可以很好的用于非活检方法的肿瘤筛查和基因型分析。
目前,百替生物在实验设计与方案执行中,应用全基因组测序、生物芯片筛选等策略。针对研究需求,制定整体化、个性化基因组学研究,与时代接轨,达到国际水平。
下一篇:干货|ncRNA数据库汇编